Cell metabolism exploration technology
WHY TRYING TO PERFORM DYNAMICAL CeLL METABOLISM EXPLORATION?
During last twenty years, technological development of large scale analysis of genes (genomics), transcripts (transcriptomics), proteins (proteomics) and metabolites (metabolomics), all referred to “omics” neologism has been thought to be the Holy Grail to find out innovative drugs. By generating
huge files of data, techniques were supposed to provide sufficient information to break gap between genotype and phenotype. Thus, it would be possible to get a holistic overview of cells global operating and even individuals networks. For that purpose, bioinformatics has developed number of methodologies to efficiently analyse, integrate and interpret “omics” data. They were thought to be able to decode complex mechanisms that could help discovery of new important therapeutic targets that direct development of specific efficient compounds addressing them. Aside Pharma and to a least extent cosmetic industry, they were supposed to be massively used in clinical laboratories to discover useful biomarkers in either pathologies diagnosis or drug management as companion tests. However, despite constant efforts and important expenses, “omics” usefulness, either used alone or combined, remains controversial. Indeed, they are still limited to research only and not yet used for daily patients’ health management.
Static versus dynamic features
Rather than watching an entire football match, looking only to its static picture, 10 minutes after its start, will never allow concluding either which team is going to win or help deciphering if the striker is in its best shape and will score a brilliant hat trick. This image holds also true for “omics” that give huge amounts of data but never clearly point out, even using complex calculations, whether system is functional or not.
Human body comprises about 40 trillion of cells, each one containing up to 6 billion proteins, the entire system being in continual changes to adapt to either internal or external changing conditions. It is well acknowledged that slight cell modifications could easily impair protein expression or activity levels thus altering global system metabolic performances leading to diseases. Moreover, some drugs affect cell function in an acute way while others may lead to cell dysfunction in a continuous manner leading on time to chronic diseases. Thus, ignoring dynamic nature over time of any biological system, alone or treated with compounds or drugs, may certainly generate bias in research.

In most high throughput “omics” studies, static quantities of one biomolecules class are measured. Transcriptomics allows semi-quantitative determination of transcript but does not give any information on their translatability. As post-transcriptional and post-translational modifications are highly regulated, no information regarding protein synthesis and activity could then be derived. It holds true for proteomics that allows quantification of large number of proteins but does not preclude on their functionality. Protein is present in a relative amount but no one knows whether it has been activated and able to work. Metabolomics share same imperfections. Measuring metabolites allow visualizing metabolic chains with intermediate and/or end-products but does not give any information on functionality of cascade enzymes.
One way to address a dynamic metabolic exploration issue should be sampling at different times. However, even if measurement iterations are performed, it presumes that quantitative measurement techniques are sufficiently precise and reproducible to be able to compare results in a dynamic fashion.
Quantification as a major issue
Among limits that hamper every “omics” to fulfil logical promises they have raised figure the constant problem of quantification. Integration of “omics” results to system biology (combining both experimental data and mathematic modelling) supposes to start with precise and accurate quantitative raw data. Indeed, whereas random biological errors could not be circumvented, introducing errors at beginning of process and accumulating such errors at each step of aggregating data will unavoidably distort mathematic model calculations. As both accuracy and precision of multi-step biological measurement procedures are always the sum of many different elementary errors resulting from either systematic or stochastic influences, at the end of such complex process integration of false results due to error accumulation would possibly generate false predictions. In a simple comparison system where large difference between two studied cases is expected, even if systematic and stochastic errors do occur, it will be still possible to draw up true concluding remarks. In a complex system in which differences can range within a few percent, no clear answer or no answer at all would be made. This adverse error propagation can be approximated by statistical calculations but unfortunately is rarely performed. If these results are sustained by economic considerations, like in the field of drug discovery, or by vital ones, in the medical field, availability of accurate quantitative methods looks like a mandatory challenge.
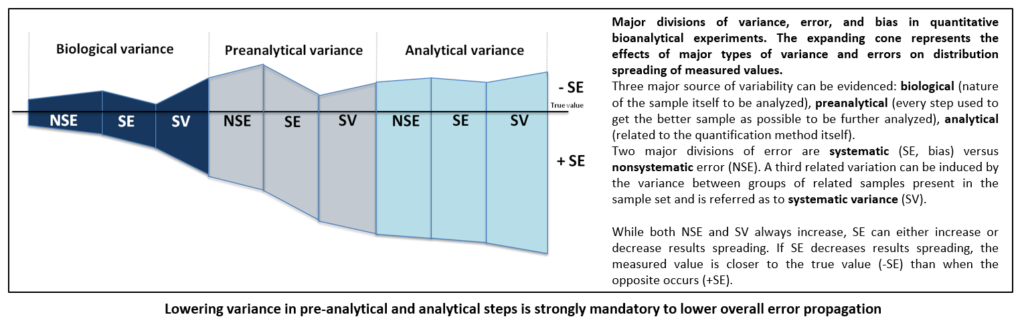
Overall variance of biological measurement can be deconvoluted into specific variances due to either nature of sample that would be analysed or methods that would be used to perform quantification. Living material is notoriously variable and most of variations are unpredictable. Thus, the experimenter has only little or absolutely no control on them. For example, it remains tricky to separate viable from non-viable cells in cell culture dish as it is also largely problematic to evidence exact percentage of tumour versus normal cells in a tissue but efforts can be made to lower these errors that may impair further interpretation. Pre-analytical and analytical phases are also very significant. As an important source of variability that can be controlled, both must be correctly mastered. It is commonly admitted that processing any measurement with bad starting sample will undoubtedly generate awful results. Many studies have evidenced, more notably in the field of clinical biology as biological results have direct impact on patients, that most of errors fall outside analytical phase. Moreover, pre-analytical steps have been found to be the most vulnerable to risk of errors. To limit their impact, recent technical recommendations regarding sampling, storage, transport and identification have been developed by consensus organizations. It holds the same in “omics” where several consortia have been constituted to recommend good practices.
Clinical biology, an example that should be followed
It has been very recently published a survey reporting that more than 70% of researchers have tried and failed to reproduce another scientist’s experiments. This is quite impossible in clinical biology as reproducibility of results is mandatory. When a patient goes in a lab for blood glucose measurement, he gets a quantitative result. If he decides to control, in another lab, his glycemia on the same blood draw, result will be the same. If he freezes his plasma and send it to a distant lab, even a foreign lab, with correct respect of pre-analytical conditions, results will be identical.
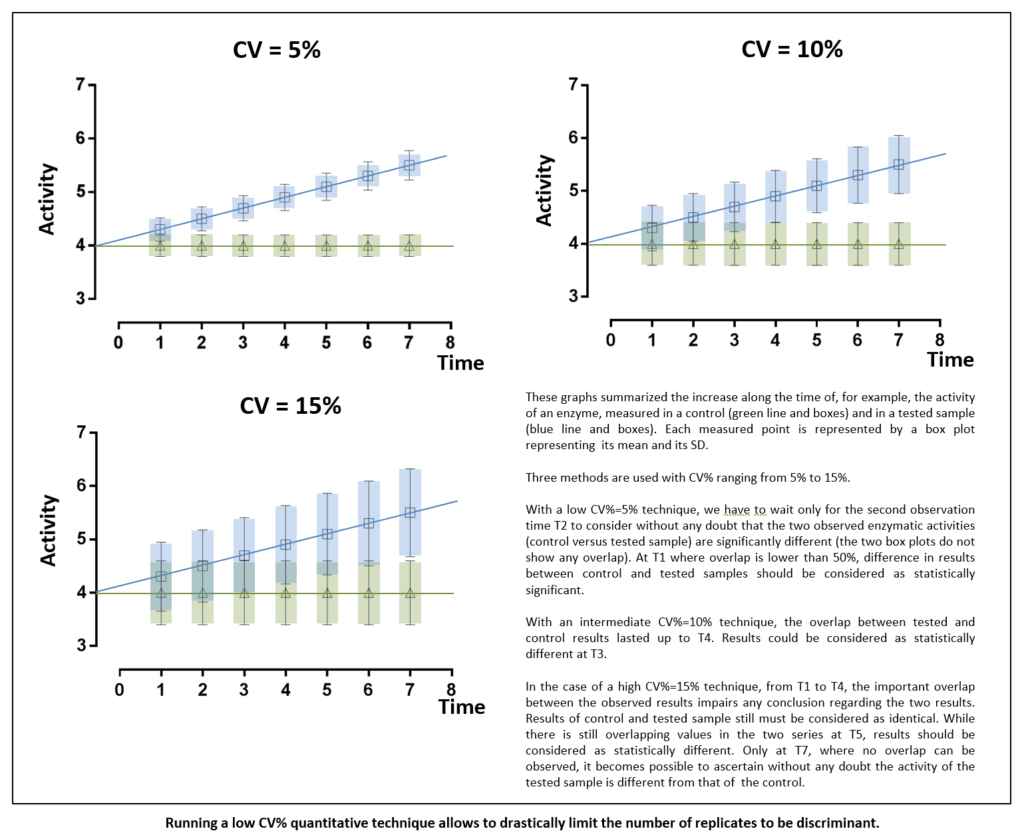
Clinical biology criteria in term of accuracy and precision are largely more stringent than research ones. The lower CV% of technique is, the less variation there is, and the highest the confidence in test results will be. Thus, being confident, there is no need to replicate test to ascertain results validity. Hence, high CV%s tests display low power to detect small-scale differences. Only low CV%s tests display sufficient power to significantly detect tiny variations. Generally, the only way to increase power with high CV%s techniques relies in increasing replicates number thus increasing starting material. Using tests with the least inherent variability will induce the least replication and thus will be the most cost-effective. Indeed, due to high precision level, experiments can be performed only once for each sample like what is routinely done for markers in patients’ plasma or serum. In every worldwide clinical biochemistry laboratories, blood glucose measurement is only performed once as CV% is largely below 1%. This procedure saves sample volumes and allows carrying out a great number of experiments on the same plasma aliquot.
.
INTEGRACELL SOLUTIONS FOR METABOLISM
CELL FUNCTIONAL EXPLORATIONS
How INTEGRACELL perform cell metabolism exploration
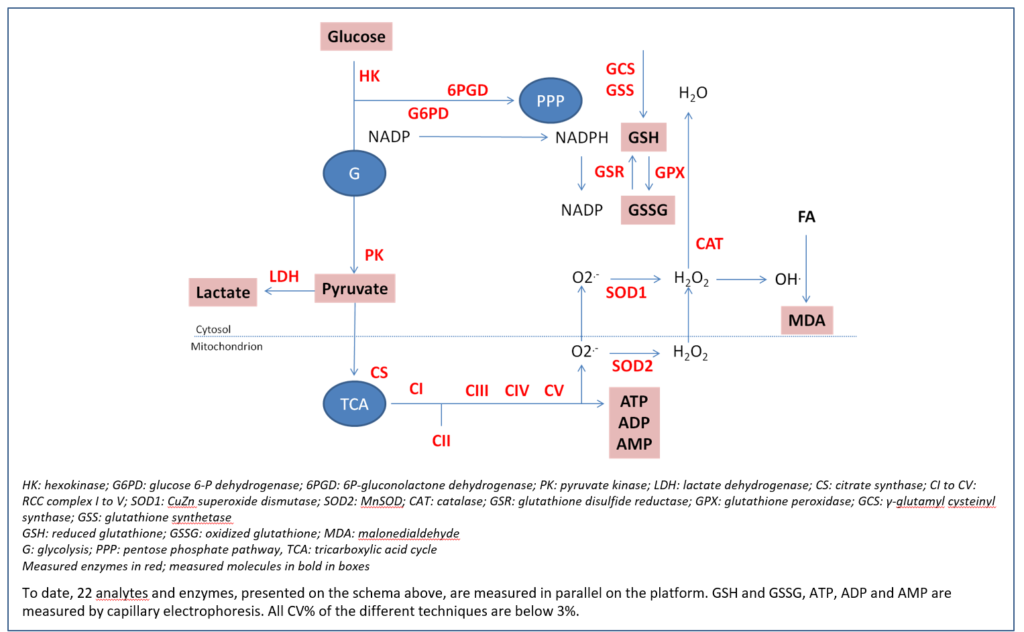
These recurrent drawbacks have prompted us to apply clinical biology criteria to our research field by diverting very precise and accurate routine clinical chemistry analyzers for parallel analysis of large number of metabolic parameters.
The apparatus, a Roche/Hitachi-ModularP analyzer, normally dedicated to routine clinical biochemistry, consists in an automated spectrophotometer that can analyze up to 300 samples in parallel and measure a maximum of 86 different photometric tests. Absorbance readings are taken every 20 s at a specified wavelength in a kinetic fashion, allowing very precise reaction follow-up. Automation allows launching one assay every 6 seconds. Thus, a multi-panel of analysis consisting on more than 20 assays may be launched in 2 minutes time limiting the biological sample evolution.

Therefore, we can assume that all measurements are performed on a same sample at the same time. Moreover, as all results are given in less than 30 minutes, it becomes possible with the same apparatus and reagents to rerun samples, whose results seem unfitting, without time degradation between series. Last, it is possible to measure in the same series either cell homogenates (as few as 3000000 cultured cells), biopsies or tissues homogenates from different species (5-10 mg required), or human plasmas, nucleated cells and red blood cells hemolysates (one 5 mL vacutainer is sufficient).

Ability to measure enzyme activities gives information not only on protein amount but also on functionality. Measurements in parallel of different parameters allow establishment of ratios (i.e enzyme activity ratios) and because CV% of each technique is below 3%, it becomes possible to compare these ratios. As it is also possible to very precisely measure protein sample concentration, all the quantitative tests can be reported to this content.
Iteration in a functional study becomes possible and allows analyzing compound or drug effects in dynamic fashion. If a time-dependent experiment has been designed, all samples are analyzed in parallel on the same apparatus at the same time with the same technical constraints, hereby limiting possible drawbacks.
Applications to functional bioenergetics
Catalytic dysfunctions in mitochondrial and/or oxidative stress pathways that lead to bioenergetics defects have been reported in large number of species (from unicellular to multicellular eukaryotes, from vegetal to animal). In mammals, whereas oxidative stress (OS) seems ubiquitously distributed, respiratory chain (RCC) dysfunction is frequently observed in specialized tissues. As compound evaluation can be performed either in preclinical or clinical stages, development of versatile diagnostic assays usable either on a large subset of cells and/or tissues or on a variety of living species (from yeast to humans) is strongly mandatory. Traditional quantification methods either for RCC or OS are usually labour-intensive and time-consuming because still largely manual. Thus, they are only available in a small number of very specialized laboratories able to extract with appropriate means mitochondria from whole cells. Most of conventional techniques lack analytical performances, especially precision, with intra- and inter-CV % largely above 10%, impairing detection of tiny variations that are common in chronic illnesses. In addition, measurement of a large panel of markers necessitates the use of a big range of different techniques. It also requires to measure parameters successively and never in parallel, generating an unavoidable degradation of starting material.
.
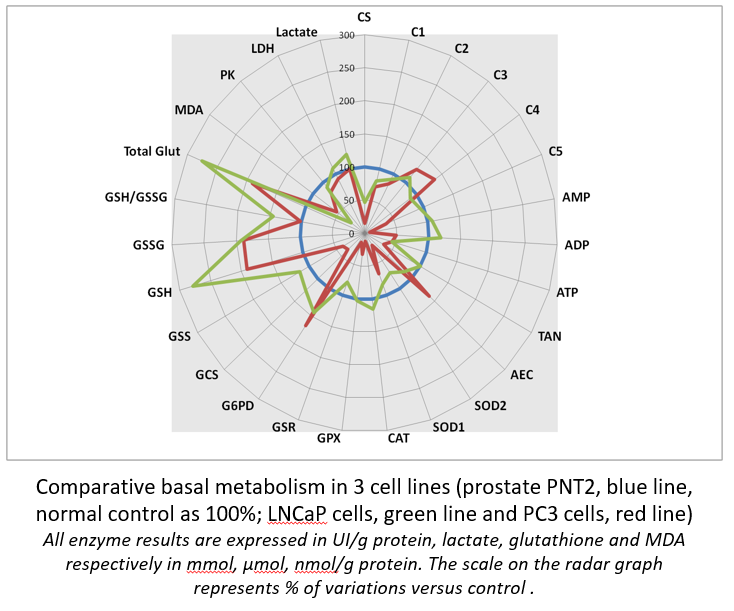
Radar panel results allows immediate comparison of different cultured cells at their metabolism level. It becomes possible to compare different cells, different tissues, different animals…
It is easy to compare a control cell line (here, normal prostate PNT2 cells) to their cancer neighbors (LNCaP and metastatic PC3 cells). Interestingly , it is of note that PC3 cell is likely to be the one that displays the most important GSH concentration, a possible way to resist to chemotherapy.
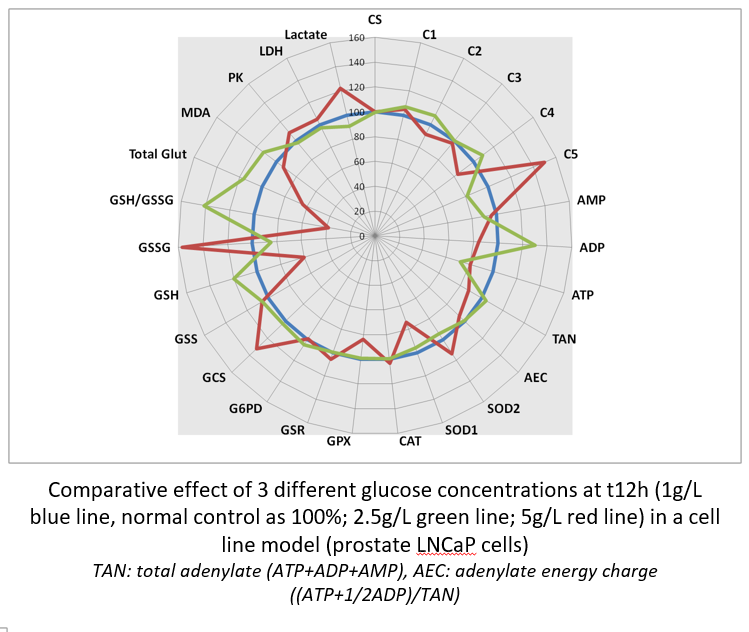
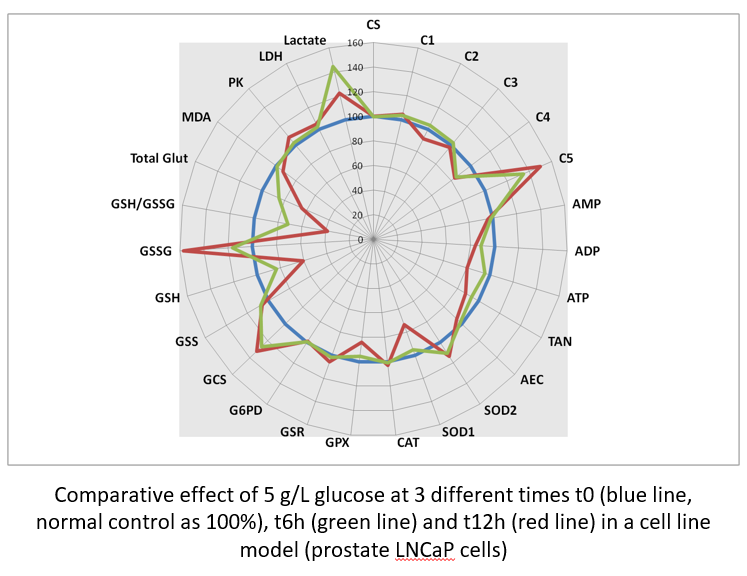
It is possible to compare the effect of drug concentrations (here, glucose as a well known toxic for cells at high concentrations). The highest glucose concentration decreases RCC with a drop in ATP synthesis that try to be compensate by an increase in complex V activity. High glucose generates an oxidative stress ,largely of mitochondrial origin (increased SOD2) ,that induces GSH depletion and GSSG increase. Dysfunction of RCC induces lactate production through LDH activity.
Aside static data, it is possible to iterate quantification and observe metabolic dynamic changes. When compared at three different times, it is possible to observe the progressive effect of high glucose on RCC dysfunction then GSH depletion and GSSG synthesis.